Search results
Search for "activation energy" in Full Text gives 102 result(s) in Beilstein Journal of Organic Chemistry.
Advancements in hydrochlorination of alkenes
- Daniel S. Müller
Beilstein J. Org. Chem. 2024, 20, 787–814, doi:10.3762/bjoc.20.72

- C–H bonds into the alkene π-bond [30]. Before reviewing polar hydrochlorination reactions in detail, it is worth mentioning several statements which were made in the Sergeev review [12]: a) The activation energy for an anti-Markovnikov addition is at least by 30 kJ mol−1 higher than for normal
- concerning the polar hydrochlorinations the activation energy for an anti-Markovnikov addition is at least by 30 kJ mol−1 higher than for normal addition. Therefore, the formation of the anti-Markovnikov product via purely cationic intermediates is never observed. The only report for the formation of the

Graphical Abstract

Scheme 1: Classes of hydrochlorination reactions discussed in this review.

Figure 1: Mayr’s nucleophilicity parameters for several alkenes. References for each compound can be consulte...

Figure 2: Hydride affinities relating to the reactivity of the corresponding alkene towards hydrochlorination....

Scheme 2: Distinction of polar hydrochlorination reactions.

Scheme 3: Reactions of styrenes with HCl gas or HCl solutions.

Figure 3: Normal temperature dependence for the hydrochlorination of (Z)-but-2-ene.

Figure 4: Pentane slows down the hydrochlorination of 11.

Scheme 4: Recently reported hydrochlorinations of styrenes with HCl gas or HCl solutions.

Scheme 5: Hydrochlorination reactions with di- and trisubstituted alkenes.

Scheme 6: Hydrochlorination of fatty acids with liquified HCl.

Scheme 7: Hydrochlorination with HCl/DMPU solutions.

Scheme 8: Hydrochlorination with HCl generated from EtOH and AcCl.

Scheme 9: Hydrochlorination with HCl generated from H2O and TMSCl.

Scheme 10: Regioisomeric mixtures of chlorooctanes as a result of hydride shifts.

Scheme 11: Regioisomeric mixtures of products as a result of methyl shifts.

Scheme 12: Applications of the Kropp procedure on a preparative scale.

Scheme 13: Curious example of polar anti-Markovnikov hydrochlorination.

Scheme 14: Unexpected and expected hydrochlorinations with AlCl3.

Figure 5: Ex situ-generated HCl gas and in situ application for the hydrochlorination of activated alkenes (*...

Scheme 15: HCl generated by Grob fragmentation of 92.

Scheme 16: Formation of chlorophosphonium complex 104 and the reaction thereof with H2O.

Scheme 17: Snyder’s hydrochlorination with stoichiometric amounts of complex 104 or 108.

Scheme 18: In situ generation of HCl by mixing of MsOH with CaCl2.

Scheme 19: First hydrochlorination of alkenes using hydrochloric acid.

Scheme 20: Visible-light-promoted hydrochlorination.

Scheme 21: Silica gel-promoted hydrochlorination of alkenes with hydrochloric acid.

Scheme 22: Hydrochlorination with hydrochloric acid promoted by acetic acid or iron trichloride.

Figure 6: Metal hydride hydrogen atom transfer reactions vs cationic reactions; BDE (bond-dissociation energy...

Scheme 23: Carreira’s first report on radical hydrochlorinations of alkenes.

Figure 7: Mechanism for the cobalt hydride hydrogen atom transfer reaction reported by Carreira.

Scheme 24: Radical “hydrogenation” of alkenes; competing chlorination reactions.

Scheme 25: Bogers iron-catalyzed radical hydrochlorination.

Scheme 26: Hydrochlorination instead of hydrogenation product.

Scheme 27: Optimization of the Boger protocol by researchers from Merck [88,89].

Figure 8: Proposed mechanism for anti-Markovnikov hydrochlorination by Nicewicz.

Scheme 28: anti-Markovnikov hydrochlorinations as reported by Nicewicz.

Figure 9: Mechanism for anti-Markovnikov hydrochlorination according to Ritter.

Scheme 29: anti-Markovnikov hydrochlorinations as reported by Nicewicz; rr (regioisomeric ratio) corresponds t...

Scheme 30: anti-Markovnikov hydrochlorinations as reported by Liu.
Enhanced host–guest interaction between [10]cycloparaphenylene ([10]CPP) and [5]CPP by cationic charges
- Eiichi Kayahara,
- Yoshiyuki Mizuhata and
- Shigeru Yamago
Beilstein J. Org. Chem. 2024, 20, 436–444, doi:10.3762/bjoc.20.38

- ). The other two isomers, 2 and 3 (Figure 5b,c), with two CPPs tilted at 15.6° and 45.5°, are 2.5 and 4.2 kJ mol−1 less stable than complex 1, respectively. The stability among the isomers is low, and the activation energy for isomerization should be very low. Therefore, all isomers are expected to be

Graphical Abstract

Figure 1: Structures of a) [10]CPP⊃C60, (b) [n+5]CPP⊃[n]CPP, and (c) [10]CPP⊃[5]CPP2+ (this work).

Figure 2: 1H NMR spectra (CD2Cl2, 25 °C) of a) a mixture of [8]–[12]CPPs and [5]CPP2+[B(C6F5)4−]2 before and aft...

Figure 3: Cyclic voltammograms of [10]CPP⊃[5]CPP2+[B(C6F5)4−]2, [10]CPP, and [5]CPP2+[B(C6F5)4−]2 in Bu4N+ B(C...

Figure 4: UV–vis–NIR spectra of [10]CPP⊃[5]CPP2+[B(C6F5)4−]2 (black), [10]CPP (blue), and [5]CPP2+[B(C6F5)4−]2...

Figure 5: Top and side view of [10]CPP⊃[5]CPP2+ complexes a) 1, b) 2, and c) 3 obtained by DFT calculation at...

Figure 6: HOMO−1, HOMO, LUMO, and LUMO+1 orbitals of [10]CPP⊃[5]CPP2+ (1), [5]CPP2+, and [10]CPP.

Figure 7: X-ray crystal structure of [10]CPP⊃[5]CPP2+[B(C6F5)4]2. a) Side and b) top views of ORTEP drawings....
Photochromic derivatives of indigo: historical overview of development, challenges and applications
- Gökhan Kaplan,
- Zeynel Seferoğlu and
- Daria V. Berdnikova
Beilstein J. Org. Chem. 2024, 20, 228–242, doi:10.3762/bjoc.20.23

- behavior was explained by the large energy gap between the ground states of the E- and Z-forms of indigo as well as low activation energy of inversion for derivatives 13. In the same year, Nielsen, Hecht and co-workers achieved a remarkable stabilization of the Z-isomer of N,N'-disubstituted indigo 24 by

Graphical Abstract

Figure 1: Precursors used in the synthesis of indigo [4].

Figure 2: a) Intramolecular (a = 2.26 Å) and intermolecular (b = 2.11 Å) hydrogen bonds in indigo, b) crystal...

Figure 3: Bond length in the indigo molecule obtained from the single crystal X-ray analysis [12], the typical bo...

Figure 4: The structure of the indigo chromophore (H-chromophore, highlighted in blue), asterisk indicates th...

Figure 5: Influence of substituents in the benzene rings on the color of indigo derivatives.

Figure 6: a) E–Z photoisomerization of indigo and b) photoinduced proton transfer in the excited state, aster...

Figure 7: Structures of indigo derivatives discussed in this review.

Figure 8: Photoswitching of N,N'-diacetylindigo (9a) in CCl4 (c = 17.1 µM; cell length = 5.0 cm) irradiated w...

Figure 9: Photoisomerization of compound 18c upon irradiation with red light and schematic representation of ...

Figure 10: Schematic representation of indigo-type (left) and amide-type (right) resonances in N,N'-acetylindi...

Figure 11: Suggested intermediates for the double bond cleavage for the thermal relaxation of N,N'-diacylindig...

Figure 12: Zwitterionic resonance structures of Z-indigo.

Figure 13: Photos of crystalline N,N'-di(Boc)indigo 17a its solutions in 1) DMSO, 2) DMF, 3) N-methyl-2-pyrrol...

Figure 14: Structural isomers of indigo.

Figure 15: Photochromism of indirubin derivatives and supramolecular complexation of the E-isomers with Schrei...

Figure 16: Photoisomerization of the protonated isoindigo.
Unraveling the role of prenyl side-chain interactions in stabilizing the secondary carbocation in the biosynthesis of variexenol B
- Moe Nakano,
- Rintaro Gemma and
- Hajime Sato
Beilstein J. Org. Chem. 2023, 19, 1503–1510, doi:10.3762/bjoc.19.107

- possibility of through-space interactions with prenyl side chains using DFT calculations. Our calculations show that (i) the unstable secondary carbocation is stabilized by the cation–π interaction from prenyl side chains, thereby lowering the activation energy, (ii) the four-membered ring formation is
- interacts with the secondary carbocation at C10, reducing the activation energy of the first step by approximately 4.7 kcal/mol. Moreover, due to the stabilization of the secondary carbocation-like intermediate IM2, the reaction proceeds stepwise rather than concertedly [7]. It was found that the final
- the stabilization of the intermediate IM2b is greater in path b, the activation energy suggests that path a is more favorable. Generally, the activation energies for terpene cyclization reactions are often below 10 kcal/mol. However, in the case of complex rearrangement reactions involving secondary

Graphical Abstract

Scheme 1: Proposed biosynthetic pathway for variexenol B.

Figure 1: (A) Results of DFT evaluation of the whole pathway of variexenol B without cation–π interaction. (B...

Figure 2: (A) Results of the DFT evaluation of the whole pathway of variexenol B including cation–π interacti...

Figure 3: (A) A representative example of the evolution of key bond lengths in the conversion of path a. (B) ...
Non-noble metal-catalyzed cross-dehydrogenation coupling (CDC) involving ether α-C(sp3)–H to construct C–C bonds
- Hui Yu and
- Feng Xu
Beilstein J. Org. Chem. 2023, 19, 1259–1288, doi:10.3762/bjoc.19.94

- and regenerates the tert-butoxyl radical to complete the entire catalytic cycle. The formation of hydrogen bonds between the oxygen of the carbonyl group and the hydrogen of the 2,2,2-trifluoroethanol (TFE) reduces the activation energy of the radical reaction and improves the coupling efficiency. In

Graphical Abstract

Scheme 1: Research progress of coupling reactions and active compounds containing α-C(sp3)-functionalized eth...

Scheme 2: Transition-metal-catalyzed CDC pathways.

Scheme 3: CDC of active methylene compounds in the α-C(sp3) position of ethers.

Scheme 4: InCl3/Cu(OTf)2/NHPI co-catalyzed CDC reaction.

Scheme 5: CDC of cyclic benzyl ethers with aldehydes.

Scheme 6: Cu-catalyzed CDC of (a) unactivated C(sp3)–H ethers with simple ketones and (b) double C(sp3)−H fun...

Scheme 7: Cu-catalyzed CDC of C(sp3)–H/C(sp3)–H bonds.

Scheme 8: Cu-catalyzed synthesis of chiral 2-substituted tetrahydropyrans.

Scheme 9: CDC of thiazole with cyclic ethers.

Scheme 10: Cu(I)-catalyzed oxidative alkenylation of simple ethers.

Scheme 11: Cross-dehydrogenation coupling of isochroman C(sp3)–H bonds with anisole C(sp2)–H bonds.

Scheme 12: Pd(OAc)2/Cu(OTf)2-catalyzed arylation of α-C(sp3)–H bonds of ethers.

Scheme 13: Cu-catalyzed C(sp3)–H/C(sp2)–H activation strategies to construct C(sp3)–C(sp2) bonds.

Scheme 14: Cu(I)-catalyzed C(sp2)–H alkylation.

Scheme 15: Cu-catalyzed C(sp3)–H/C(sp)–H activation to construct C(sp3)–C(sp) bonds (H2BIP: 2,6-bis(benzimidaz...

Scheme 16: Fe-catalyzed CDC reaction pathways.

Scheme 17: Fe2(CO)9-catalyzed functionalization of C–H bonds.

Scheme 18: Ligand-promoted Fe-catalyzed CDC reaction of N-methylaniline with ethers.

Scheme 19: Fe-catalyzed CDC of C(sp3)–H/C(sp3)–H bonds.

Scheme 20: Fe-catalyzed hydroalkylation of α,β-unsaturated ketones with ethers.

Scheme 21: Solvent-free Fe(NO3)3-catalyzed CDC of C(sp3)–H/C(sp2)–H bonds.

Scheme 22: Alkylation of disulfide compounds to afford tetrasubstituted alkenes.

Scheme 23: Fe-catalyzed formation of 1,1-bis-indolylmethane derivatives.

Scheme 24: Alkylation of coumarins and flavonoids.

Scheme 25: Direct CDC α-arylation of azoles with ethers.

Scheme 26: CDC of terminal alkynes with C(sp3)–H bonds adjacent to oxygen, sulfur or nitrogen atoms.

Scheme 27: Alkylation of terminal alkynes.

Scheme 28: Co-catalyzed functionalization of glycine esters.

Scheme 29: Co-catalyzed construction of C(sp2)–C(sp3) bonds.

Scheme 30: Co-catalyzed CDC of imidazo[1,2-a]pyridines with isochroman.

Scheme 31: Co-catalyzed C–H alkylation of (benz)oxazoles with ethers.

Scheme 32: Cobalt-catalyzed CDC between unactivated C(sp2)–H and C(sp3)–H bonds.

Scheme 33: MnO2-catalyzed CDC of the inactive C(sp3)-H.

Scheme 34: Oxidative cross-coupling of ethers with enamides.

Scheme 35: Ni(II)-catalyzed CDC of indoles with 1,4-dioxane.

Scheme 36: Chemo- and regioselective ortho- or para-alkylation of pyridines.

Scheme 37: Asymmetric CDC of 3,6-dihydro-2H-pyrans with aldehydes.

Scheme 38: CDC of heterocyclic aromatics with ethers.

Scheme 39: Indium-catalyzed alkylation of DHPs with 1,3-dicarbonyl compounds.

Scheme 40: Rare earth-metal-catalyzed CDC reaction.

Scheme 41: Visible-light-driven CDC of cycloalkanes with benzazoles.

Scheme 42: Photoinduced alkylation of quinoline with cyclic ethers.

Scheme 43: Photocatalyzed CDC reactions between α-C(sp3)–H bonds of ethers and C(sp2)–H bonds of aromatics.
Exploring the role of halogen bonding in iodonium ylides: insights into unexpected reactivity and reaction control
- Carlee A. Montgomery and
- Graham K. Murphy
Beilstein J. Org. Chem. 2023, 19, 1171–1190, doi:10.3762/bjoc.19.86

- ]. Computational investigations were conducted to better understand these reactions, and it was determined that the changing alkyl motif (e.g., dimethyl, cyclopentyl, adamantyl) had minimal impact on the activation energy of the fluorination reactions. The reaction coordinate was calculated for Meldrum’s acid
- comparing ylides 31 and 70 in this reductive elimination step, which suggested that the ortho-nitro group of 72 (and 71, by extension) played a role other than lowering the activation energy of this step. Given that other optimized halogen- and hydrogen-bonded conformations were found between thioamide and
- . Proposed mechanism of the formal C–H insertion of pyrrole under blue LED irradiation. Mechanistic proposal for the X–H insertion reactions of iodonium ylides. Calculated reaction coordinate for the radiofluorination of iodonium ylide 60. Difference in Gibbs activation energy for ortho- or para-anisyl

Graphical Abstract

Figure 1: Generic representation of halogen bonding.

Figure 2: Quantitative evaluation of σ-holes in monovalent iodine-containing compounds; and, qualitative mole...

Figure 3: Quantitative evaluation of σ-holes in hypervalent iodine-containing molecules; and, qualitative MEP...

Figure 4: Quantitative evaluation of σ-holes in iodonium ylides; and, qualitative MEP map of I-12 from −0.083...

Scheme 1: Outline of possible reaction pathways between iodonium ylides and Lewis basic nucleophiles (top); a...

Scheme 2: Metal-free cyclopropanations of iodonium ylides, either as intermolecular (a) or intramolecular pro...

Figure 5: Zwitterionic mechanism for intramolecular cyclopropanation of iodonium ylides (left); and, stepwise...

Scheme 3: Metal-free intramolecular cyclopropanation of iodonium ylides.

Figure 6: Concerted cycloaddition pathway for the metal-free, intramolecular cyclopropanation of iodonium yli...

Scheme 4: Reaction of ylide 6 with diphenylketene to form lactone 24 and 25.

Figure 7: Nucleophilic (top) and electrophilic (bottom) addition pathways proposed by Koser and Hadjiarapoglo...

Scheme 5: Indoline synthesis from acyclic iodonium ylide 31 and tertiary amines.

Scheme 6: N-Heterocycle synthesis from acyclic iodonium ylide 31 and secondary amines.

Figure 8: Proposed mechanism for the formation of 33a from iodonium ylides and amines, involving an initial h...

Scheme 7: Indoline synthesis from acyclic iodonium ylides 39 and tertiary amines under blue light photocataly...

Scheme 8: Metal-free cycloproponation of iodonium ylides under blue LED irradiation. aUsing trans-β-methylsty...

Figure 9: Proposed mechanism of the cyclopropanation between iodonium ylides and alkenes under blue LED irrad...

Scheme 9: Formal C–H alkylation of iodonium ylides by nucleophilic heterocycles under blue LED irradiation.

Figure 10: Proposed mechanism of the formal C–H insertion of pyrrole under blue LED irradiation.

Scheme 10: X–H insertions between iodonium ylides and carboxylic acids, phenols and thiophenols.

Figure 11: Mechanistic proposal for the X–H insertion reactions of iodonium ylides.

Scheme 11: Radiofluorination of biphenyl using iodonium ylides 54a–e derived from various β-dicarbonyl auxilia...

Scheme 12: Radiofluorination of arenes using spirocycle-derived iodonium ylides 56.

Scheme 13: Radiofluorination of arenes using SPIAd-derived iodonium ylides 58.

Figure 12: Calculated reaction coordinate for the radiofluorination of iodonium ylide 60.

Scheme 14: Radiofluorination of iodonium ylides possessing various ortho- and para-substituents on the iodoare...

Figure 13: Difference in Gibbs activation energy for ortho- or para-anisyl derived iodonium ylides 63a and 63b....

Figure 14: Proposed equilibration of intermediates to transit between 64a (the initial adduct formed between 6...

Scheme 15: Comparison of 31 and ortho-methoxy iodonium ylide 39 in rhodium-catalyzed cyclopropanation and cycl...

Figure 15: X-ray crystal structure of dimeric 39 [6], (CCDC# 893474) [143,144].

Scheme 16: Enaminone synthesis using diazonium and iodonium ylides.

Figure 16: Transition state calculations for enaminone synthesis from iodonium ylides and thioamides.

Scheme 17: The reaction between ylides 73a–f and N-methylpyrrole under 365 nm UV irradiation.

Figure 17: Crystal structures of 76c (top) and 76e (bottom) [101], (CCDC# 2104180 & 2104181) [143,144].
Construction of hexabenzocoronene-based chiral nanographenes
- Ranran Li,
- Di Wang,
- Shengtao Li and
- Peng An
Beilstein J. Org. Chem. 2023, 19, 736–751, doi:10.3762/bjoc.19.54

- Gibbs activation energy of enantiomer 53 for the racemization process was determined as 33.0 kcal mol−1 at 298 K. The CPL spectra of M-53 and P-53 showed an emission maximum at 560 nm with glum value of 2.3 × 10−4. Instead of helicene formation in the final Scholl-type ring formation step, Martín and co

Graphical Abstract

Scheme 1: Construction of HBC by Scholl reaction from hexaphenylbenzene.

Scheme 2: Synthesis of seco-HBC-based chiral nanographenes.

Scheme 3: Synthesis of nitrogen-doped, seco-HBC-based chiral nanographenes.

Scheme 4: Synthesis of π-extended [7]- and [9]helicene containing chiral nanographenes.

Scheme 5: Synthesis of “HBC-dimer”-based chiral nanographenes.

Scheme 6: Synthesis of “HBC-dimer”-based chiral nanographenes.

Scheme 7: Synthesis of axis-based chiral nanographenes.

Scheme 8: Synthesis of “HBC-trimers”-based nanoribbons.

Scheme 9: Synthesis of “HBC-trimers”-based, triangle-shaped chiral nanographenes.

Scheme 10: Synthesis of “HBC-trimers”-based, triangle-shaped chiral nanographenes.

Scheme 11: Synthesis of HBC-based multilayer nanographenes.

Scheme 12: Synthesis of a chiral nanographene constructed by “HBC-tetramers”.

Scheme 13: Synthesis of a triskelion-shaped nanographene constructed by four HBCs.

Scheme 14: Synthesis of a three-dimensional nanographene bearing four HBCs.

Scheme 15: Synthesis of a chiral nanographene constructed by five HBC units.

Scheme 16: Synthesis of a chiral nanographene constructed by seven HBC units.
Computational studies of Brønsted acid-catalyzed transannular cycloadditions of cycloalkenone hydrazones
- Manuel Pedrón,
- Jana Sendra,
- Irene Ginés,
- Tomás Tejero,
- Jose L. Vicario and
- Pedro Merino
Beilstein J. Org. Chem. 2023, 19, 477–486, doi:10.3762/bjoc.19.37

- model [30] the distortion energy (ΔE≠d) corresponds to the difference between the single point corresponding to interacting 3 and 4, and the sum of single-point calculations for TS2-a and TS2b. The apparent activation energy (ΔE≠app) refers to the energy difference between TS2 and the interacting

Graphical Abstract

Scheme 1: Experimental data (series a–d, k) and non-studied examples (series e–j) for transannular cycloaddit...

Figure 1: Optimized (m062x/6-31G(d)) geometries for the transition structures of series a–f.

Figure 2: Top: Cycloaddition of protonated hydrazones as inverse-demand reaction of cycloaddition of azomethi...

Figure 3: Global electron density transfer (GEDT). Dashed black line indicates both TS.

Figure 4: ELF analysis for the reaction of series b leading to a system 6-6. Black trace corresponds to IRC. ...

Figure 5: Quantitative NCI analysis [36] for the reaction of series a–f leading to fused cyclohexanes. The result...

Figure 6: (a) Transannular cycloadditons of compounds 1a–k. (b) Houk’s distortion model applied to the reacti...

Scheme 2: Reaction with simple models.
B–N/B–H Transborylation: borane-catalysed nitrile hydroboration
- Filip Meger,
- Alexander C. W. Kwok,
- Franziska Gilch,
- Dominic R. Willcox,
- Alex J. Hendy,
- Kieran Nicholson,
- Andrew D. Bage,
- Thomas Langer,
- Thomas A. Hunt and
- Stephen P. Thomas
Beilstein J. Org. Chem. 2022, 18, 1332–1337, doi:10.3762/bjoc.18.138

- activation energy supported the proposed routes (see Supporting Information File 1 for details). Conclusion In summary, a borane-catalysed hydroboration of nitriles to give primary amines has been developed, transforming the previously stoichiometric reagent H3B·SMe2 into a catalyst. B-N/B–H transborylation

Graphical Abstract

Scheme 1: a) Derivatives of primary amines in materials chemistry, pharmaceuticals, and agrochemicals; b) thi...

Scheme 2: Substrate scope of borane-catalysed nitrile hydroboration with HBpin. Conditions: nitrile (0.50 mmo...

Scheme 3: a) Proposed mechanism; b) H-B-9-BBN-catalysed heptanenitrile hydroboration (yield determined by 1H ...
Polymer and small molecule mechanochemistry: closer than ever
- José G. Hernández
Beilstein J. Org. Chem. 2022, 18, 1225–1235, doi:10.3762/bjoc.18.128

- is observed [46]. This difference is believed to be related to the exertion of tensile forces along the glycosidic linkage of the polymer chain during ball milling, which may lower the activation energy for the depolymerization of chitin. Indeed, DFT calculations using the N-acetylglucosamine dimer
- as the model compound showed that the application of pulling forces to selected atoms in the dimer perturb the reaction, making the depolymerization easier to occur [45]. In contrast, no change in the activation energy of the deacetylation step was observed with the introduction of the pulling forces
- . The decrease in the activation energy for the mechanochemical depolymerization of chitin was attributed to force-induced conformational changes in the structure, which destabilize the reactant state upon the introduction of a sufficient pulling force (Figure 3). Evidently, ball milling techniques

Graphical Abstract

Figure 1: Representation of (a) cavitation and elongational flow caused by pulsed ultrasonication, (b) mixer ...

Scheme 1: (a) Mechanochemical activation of anthracene–endoperoxide mechanophore incorporated in the cross-li...

Scheme 2: Mechanochemical activation of dendronized polymer-based compound 4 by ultrasonication and ball mill...

Figure 2: Structure of cellulose and chitin and approximation to the structure of lignin.

Figure 3: Tensile forces by ball milling change the conformation of a chitin model compound. This deformation...

Figure 4: (a) Representation of a collision between the ball and a particle of a chitin sample and (b) mechan...

Figure 5: (a) Ultrasound-induced ATRP using piezoelectric BaTiO3 and (b) mechanochemical atom transfer radica...

Figure 6: Mechanochemical solid-state complexation of organic capsule 5 with fullerenes C70 in a planetary ba...

Scheme 3: Comparative mechanochemical dissociation of the central C–C bond in TASN derivatives 6 and 8.
Experimental and theoretical studies on the synthesis of 1,4,5-trisubstituted pyrrolidine-2,3-diones
- Nguyen Tran Nguyen,
- Vo Viet Dai,
- Nguyen Ngoc Tri,
- Luc Van Meervelt,
- Nguyen Tien Trung and
- Wim Dehaen
Beilstein J. Org. Chem. 2022, 18, 1140–1153, doi:10.3762/bjoc.18.118

- with an activation energy of about 10.7 kcal·mol−1. On the other hand, if the removal of one H2O molecule occurs from the O–H and N–H groups, IS5 transforms into IS9 through TS14 with a high barrier of 31.8 kcal·mol−1. IS9 is then converted to product 10ab based on the shifting of H atom from the O–H
- bond to N atom (Scheme 4). This process is thermodynamically favorable by a Gibbs free energy (ΔG) of −6.5 kcal·mol−1. In addition, a second way from 4a, through the TS5 transition state, to form the IS3 intermediate requires an activation energy of ca. 33.7 kcal·mol−1. IS3 could release one H2O
- IS8 can be omitted because it is so fast with a small activation energy of 2.1 kcal·mol−1), the formation of product 10ab is superior as compared to its isomer. In terms of the value of the rate constant k, the direction of 10ab formation is in the range of 103–106 times faster than 10ab-v2. The

Graphical Abstract

Figure 1: Structure of naturally occurring and synthetic 2-pyrrolidone derivatives.

Figure 2: Structure of natural compounds containing a 1,5-dihydro-2H-pyrrol-2-one subunit.

Scheme 1: Synthesis of substituted 4-acetyl-3-hydroxy-3-pyrroline-2-ones 4 via three-component reaction.

Scheme 2: Proposed mechanistic path for the synthesis of substituted 4-acetyl-3-hydroxy-3-pyrroline-2-ones.

Scheme 3: Tautomerism of compounds 4a–c in DMSO.

Figure 3: View of the molecular structure of compound 10aa with atom labeling. Displacement ellipsoids are dr...

Figure 4: The PES of reaction for the synthesis of 1,4,5-trisubstituted pyrrolidine-2,3-dione 10ab enamine de...

Scheme 4: Reaction pathways from 4a, 4a’ to 10ab via IS5 in the gase phase.

Scheme 5: Reaction pathways from 4a to 10ab-v2 via IS3 in the gase phase.

Figure 5: The PES for the possible pathways (1), (2), (3), and (4) in ethanol solvent.

Figure 6: The optimized structures of some reactants, intermediates, transition states, and products in the p...
Comparative study of thermally activated delayed fluorescent properties of donor–acceptor and donor–acceptor–donor architectures based on phenoxazine and dibenzo[a,j]phenazine
- Saika Izumi,
- Prasannamani Govindharaj,
- Anna Drewniak,
- Paola Zimmermann Crocomo,
- Satoshi Minakata,
- Leonardo Evaristo de Sousa,
- Piotr de Silva,
- Przemyslaw Data and
- Youhei Takeda
Beilstein J. Org. Chem. 2022, 18, 459–468, doi:10.3762/bjoc.18.48

- in electron density on the acceptor and the electron-donating power of POZ. Therefore, gradual increase of electron-donating strength brings T1 energy closer to the acceptor T1 energy and leads to a smaller EST gap. But, the activation energy Ea for the DF process, which was calculated from the
- Arrhenius plot obtained from the increase of the DF intensity against temperature, was lower for 1 (Ea = 27 meV) when compared to POZ-DBPHZ (Ea = 47 meV, Table 2) in Zeonex®. The directly determined activation energy of the D–A-type compound is half than that of the D–A–D compound, which is in contradiction
- to the ΔEST value (Table 2). If we support the observation with the DF/PF results that present a stronger TADF property for the mono-substituted derivative 1, the conclusion of misleading ΔEST comparison can be reached. To avoid confusion, a more effective way is to compare only the activation energy

Graphical Abstract

Figure 1: Chemical structures of 1 and POZ-DBPHZ.

Scheme 1: Synthesis of compound 1.

Figure 2: Steady-state UV–vis absorption (Abs) and photoluminescence (PL) spectra of dilute solutions (c ≈ 10...

Figure 3: Time-resolved PL decay profiles (intensity vs delay time) and spectra of 1 in a), b) Zeonex® and c,...

Figure 4: The characteristics of the OLED devices: a) electroluminescence spectra; b) current density-bias ch...

Figure 5: Schematics of the TADF mechanisms along with NTOs for the relevant electronic states for a) D–A com...
Iridium-catalyzed hydroacylation reactions of C1-substituted oxabenzonorbornadienes with salicylaldehyde: an experimental and computational study
- Angel Ho,
- Austin Pounder,
- Krish Valluru,
- Leanne D. Chen and
- William Tam
Beilstein J. Org. Chem. 2022, 18, 251–261, doi:10.3762/bjoc.18.30

- hypothesized the reductive elimination step was the RDS for rhodium-catalyzed intramolecular hydroacylation reactions [74]. Based on the activation energy of the reverse step of reductive elimination 3TS2 (37.4–41.4 kcal/mol), we predict reductive elimination, and subsequent C–C formation, to be irreversible
- reductive elimination transition states, 2bTS3b is the more energetically accessible transition state with an energy barrier of 5.9 kcal/mol, that is 4.9 kcal/mol lower in energy compared to 2aTS3a. This large difference in activation energy (ΔΔG‡) between the two competing transition states offers
- an activation energy of 4.8 to 5.6 kcal/mol, respectively, to produce the aforementioned thermodynamically stable Ir(I) alkoxide intermediates INa3 and INb3. Based on the extremely high energy barrier required for acyl migration over hydride migration, we hypothesize iridium-catalyzed hydroacylation

Graphical Abstract

Scheme 1: Previously reported metal-catalyzed reactions of heterobicyclic alkenes and applications towards th...

Scheme 2: Iridium-catalyzed hydroacylation of C1-substituted OBDs 13a–k with salicylaldehyde 14.

Scheme 3: Competition reaction of different C1-substituted OBDs.

Figure 1: Potential energy profile of the PCM solvation model for the hydrometalation/reductive elimination p...

Figure 2: Potential energy profile of the PCM solvation model for the carbometalation/reductive elimination p...

Figure 3: Potential energy profile of the PCM solvation model for the endo hydrometalation/reductive eliminat...

Figure 4: Potential energy profile of the PCM solvation model for the Ir/diene-catalyzed hydroacylation of Me...
Iron-catalyzed domino coupling reactions of π-systems
- Austin Pounder and
- William Tam
Beilstein J. Org. Chem. 2021, 17, 2848–2893, doi:10.3762/bjoc.17.196


Graphical Abstract

Figure 1: Price comparison among iron and other transition metals used in catalysis.

Scheme 1: Typical modes of C–C bond formation.

Scheme 2: The components of an iron-catalyzed domino reaction.

Scheme 3: Iron-catalyzed tandem cyclization and cross-coupling reactions of iodoalkanes 1 with aryl Grignard ...

Scheme 4: Three component iron-catalyzed dicarbofunctionalization of vinyl cyclopropanes 14.

Scheme 5: Three-component iron-catalyzed dicarbofunctionalization of alkenes 21.

Scheme 6: Double carbomagnesiation of internal alkynes 31 with alkyl Grignard reagents 32.

Scheme 7: Iron-catalyzed cycloisomerization/cross-coupling of enyne derivatives 35 with alkyl Grignard reagen...

Scheme 8: Iron-catalyzed spirocyclization/cross-coupling cascade.

Scheme 9: Iron-catalyzed alkenylboration of alkenes 50.

Scheme 10: N-Alkyl–N-aryl acrylamide 60 CDC cyclization with C(sp3)–H bonds adjacent to a heteroatom.

Scheme 11: 1,2-Carboacylation of activated alkenes 60 with aldehydes 65 and alcohols 67.

Scheme 12: Iron-catalyzed dicarbonylation of activated alkenes 68 with alcohols 67.

Scheme 13: Iron-catalyzed cyanoalkylation/radical dearomatization of acrylamides 75.

Scheme 14: Synergistic photoredox/iron-catalyzed 1,2-dialkylation of alkenes 82 with common alkanes 83 and 1,3...

Scheme 15: Iron-catalyzed oxidative coupling/cyclization of phenol derivatives 86 and alkenes 87.

Scheme 16: Iron-catalyzed carbosulfonylation of activated alkenes 60.

Scheme 17: Iron-catalyzed oxidative spirocyclization of N-arylpropiolamides 91 with silanes 92 and tert-butyl ...

Scheme 18: Iron-catalyzed free radical cascade difunctionalization of unsaturated benzamides 94 with silanes 92...

Scheme 19: Iron-catalyzed cyclization of olefinic dicarbonyl compounds 97 and 100 with C(sp3)–H bonds.

Scheme 20: Radical difunctionalization of o-vinylanilides 102 with ketones and esters 103.

Scheme 21: Dehydrogenative 1,2-carboamination of alkenes 82 with alkyl nitriles 76 and amines 105.

Scheme 22: Iron-catalyzed intermolecular 1,2-difunctionalization of conjugated alkenes 107 with silanes 92 and...

Scheme 23: Four-component radical difunctionalization of chemically distinct alkenes 114/115 with aldehydes 65...

Scheme 24: Iron-catalyzed carbocarbonylation of activated alkenes 60 with carbazates 117.

Scheme 25: Iron-catalyzed radical 6-endo cyclization of dienes 119 with carbazates 117.

Scheme 26: Iron-catalyzed decarboxylative synthesis of functionalized oxindoles 130 with tert-butyl peresters ...

Scheme 27: Iron‑catalyzed decarboxylative alkylation/cyclization of cinnamamides 131/134.

Scheme 28: Iron-catalyzed carbochloromethylation of activated alkenes 60.

Scheme 29: Iron-catalyzed trifluoromethylation of dienes 142.

Scheme 30: Iron-catalyzed, silver-mediated arylalkylation of conjugated alkenes 115.

Scheme 31: Iron-catalyzed three-component carboazidation of conjugated alkenes 115 with alkanes 101/139b and t...

Scheme 32: Iron-catalyzed carboazidation of alkenes 82 and alkynes 160 with iodoalkanes 20 and trimethylsilyl ...

Scheme 33: Iron-catalyzed asymmetric carboazidation of styrene derivatives 115.

Scheme 34: Iron-catalyzed carboamination of conjugated alkenes 115 with alkyl diacyl peroxides 163 and acetoni...

Scheme 35: Iron-catalyzed carboamination using oxime esters 165 and arenes 166.

Scheme 36: Iron-catalyzed iminyl radical-triggered [5 + 2] and [5 + 1] annulation reactions with oxime esters ...

Scheme 37: Iron-catalyzed decarboxylative alkyl etherification of alkenes 108 with alcohols 67 and aliphatic a...

Scheme 38: Iron-catalyzed inter-/intramolecular alkylative cyclization of carboxylic acid and alcohol-tethered...

Scheme 39: Iron-catalyzed intermolecular trifluoromethyl-acyloxylation of styrene derivatives 115.

Scheme 40: Iron-catalyzed carboiodination of terminal alkenes and alkynes 180.

Scheme 41: Copper/iron-cocatalyzed cascade perfluoroalkylation/cyclization of 1,6-enynes 183/185.

Scheme 42: Iron-catalyzed stereoselective carbosilylation of internal alkynes 187.

Scheme 43: Synergistic photoredox/iron catalyzed difluoroalkylation–thiolation of alkenes 82.

Scheme 44: Iron-catalyzed three-component aminoazidation of alkenes 82.

Scheme 45: Iron-catalyzed intra-/intermolecular aminoazidation of alkenes 194.

Scheme 46: Stereoselective iron-catalyzed oxyazidation of enamides 196 using hypervalent iodine reagents 197.

Scheme 47: Iron-catalyzed aminooxygenation for the synthesis of unprotected amino alcohols 200.

Scheme 48: Iron-catalyzed intramolecular aminofluorination of alkenes 209.

Scheme 49: Iron-catalyzed intramolecular aminochlorination and aminobromination of alkenes 209.

Scheme 50: Iron-catalyzed intermolecular aminofluorination of alkenes 82.

Scheme 51: Iron-catalyzed aminochlorination of alkenes 82.

Scheme 52: Iron-catalyzed phosphinoylazidation of alkenes 108.

Scheme 53: Synergistic photoredox/iron-catalyzed three-component aminoselenation of trisubstituted alkenes 82.
Fritsch–Buttenberg–Wiechell rearrangement of magnesium alkylidene carbenoids leading to the formation of alkynes
- Tsutomu Kimura,
- Koto Sekiguchi,
- Akane Ando and
- Aki Imafuji
Beilstein J. Org. Chem. 2021, 17, 1352–1359, doi:10.3762/bjoc.17.94

- ), and the C1–Cl bond (C1···Cl: 3.22 Å) was cleaved. The activation energy for this reaction was estimated to be 14.9 kcal/mol. The IRC calculation revealed that the chlorine atom gradually dissociated from the carbenoid carbon atom as the phenyl group approached the carbenoid carbon atom [38][39]. Then

Graphical Abstract

Scheme 1: Synthesis of alkynes from carbonyl compounds through one-carbon homologation.

Scheme 2: Reactions of magnesium alkylidene carbenoids 3, generated from sulfoxides 2 and iPrMgCl.

Scheme 3: Synthesis of sulfoxides 2 and 5–8 from carbonyl compounds 1.

Scheme 4: Reaction of sulfoxides 5 and 6 with isopropylmagnesium chloride.

Scheme 5: Reaction of sulfoxide 2c with isopropylmagnesium chloride.

Scheme 6: Reaction of 13C-labeled sulfoxides [13C]-(E)-2e and [13C]-(Z)-2e with iPrMgCl.

Scheme 7: A plausible reaction mechanism for the formation of alkynes 4. a) 1,2-Rearrangement readily takes p...

Figure 1: Optimized geometries of reactant (E)-3e, transition state (E)-3e‡, and product 4e·MgCl2 for the FBW...
Valorisation of plastic waste via metal-catalysed depolymerisation
- Francesca Liguori,
- Carmen Moreno-Marrodán and
- Pierluigi Barbaro
Beilstein J. Org. Chem. 2021, 17, 589–621, doi:10.3762/bjoc.17.53

- °C provided a hydrocarbon oil in 92% yield, 71.4% of which were attributable to styrene monomer [156]. A decrease of 56 kJ mol−1 for the activation energy of PS depolymerisation was calculated in the presence of the catalysts. More recently, high-porosity montmorillonite (Mt) was used to prepare Mg

Graphical Abstract

Figure 1: Potential classification of plastic recycling processes. The area covered by the present review is ...

Figure 2: EG produced during glycolytic depolymerisation of PET using DEG + DPG as solvent and titanium(IV) n...

Scheme 1: Simplified representation of the conversion of 1,4-PBD to C16–C44 macrocycles using Ru metathesis c...

Figure 3: Main added-value monomers obtainable by catalytic depolymerisation of PET via chemolytic methods.

Scheme 2: Hydrogenolytic depolymerisation of PET by ruthenium complexes.

Scheme 3: Depolymerisation of PET via catalytic hydrosilylation by Ir(III) pincer complex.

Scheme 4: Catalytic hydrolysis (top) and methanolysis (bottom) reactions of PET.

Scheme 5: Depolymerisation of PET by glycolysis with ethylene glycol.

Figure 4: Glycolysis of PET: evolution of BHET yield over time, with and without zinc acetate catalyst (196 °...

Scheme 6: Potential activated complex for the glycolysis reaction of PET catalysed by metallated ILs and evol...

Scheme 7: One-pot, two-step process for PET repurposing via chemical recycling.

Scheme 8: Synthetic routes to PLA.

Scheme 9: Structures of the zinc molecular catalysts used for PLA-methanolysis in various works. a) See [265], b) ...

Scheme 10: Depolymerisation of PLLA by Zn–N-heterocyclic carbene complex.

Scheme 11: Salalen ligands.

Scheme 12: Catalytic hydrogenolysis of PLA.

Scheme 13: Catalytic hydrosilylation of PLA.

Scheme 14: Hydrogenative depolymerisation of PBT and PCL by molecular Ru catalysts.

Scheme 15: Glycolysis reaction of PCT by diethylene glycol.

Scheme 16: Polymerisation–depolymerisation cycle of 3,4-T6GBL.

Scheme 17: Polymerisation–depolymerisation cycle of 2,3-HDB.

Scheme 18: Hydrogenative depolymerisation of PBPAC by molecular Ru catalysts.

Scheme 19: Catalytic hydrolysis (top), alcoholysis (middle) and aminolysis (bottom) reactions of PBPAC.

Scheme 20: Hydrogenative depolymerisation of PPC (top) and PEC (bottom) by molecular Ru catalysts.

Scheme 21: Polymerisation-depolymerisation cycle of BEP.

Scheme 22: Hydrogenolysis of polyamides using soluble Ru catalysts.

Scheme 23: Catalytic depolymerisation of epoxy resin/carbon fibres composite.

Scheme 24: Depolymerisation of polyethers with metal salt catalysts and acyl chlorides.

Scheme 25: Proposed mechanism for the iron-catalysed depolymerisation reaction of polyethers. Adapted with per...
CF3-substituted carbocations: underexploited intermediates with great potential in modern synthetic chemistry
- Anthony J. Fernandes,
- Armen Panossian,
- Bastien Michelet,
- Agnès Martin-Mingot,
- Frédéric R. Leroux and
- Sébastien Thibaudeau
Beilstein J. Org. Chem. 2021, 17, 343–378, doi:10.3762/bjoc.17.32

- analogue (dC–O = 1.438 Å) and strongly inhibits the formation of the α-(trifluoromethyl)bisarylcarbenium ion, as illustrated by the higher activation energy needed for the dehydration (ΔECF3 = 21.0 kcal⋅mol−1 vs ΔECH3 = 14.8 kcal⋅mol−1 at the B3LYP/6-31+G(d,p) level). On the other hand, the first arylation

Graphical Abstract

Figure 1: Stabilizing interaction in the CF3CH2+ carbenium ion (top) and structure of the first observable fl...

Scheme 1: Isodesmic equations accounting for the destabilizing effect of the CF3 group. ΔE in kcal⋅mol−1, cal...

Scheme 2: Stabilizing effect of fluorine atoms by resonance electron donation in carbenium ions (δ in ppm).

Scheme 3: Direct in situ NMR observation of α-(trifluoromethyl)carbenium ion or protonated alcohols. Δδ = δ19...

Scheme 4: Reported 13C NMR chemical shifts for the α-(trifluoromethyl)carbenium ion 10c (δ in ppm).

Scheme 5: Direct NMR observation of α-(trifluoromethyl)carbenium ions in situ (δ in ppm).

Scheme 6: Illustration of the ion pair solvolysis mechanism for sulfonate 13f. YOH = solvent.

Figure 2: Solvolysis rate for 13a–i and 17.

Figure 3: Structures of allyl triflates 18 and 19 and allyl brosylate 20. Bs = p-BrC6H4SO2.

Figure 4: Structure of tosylate derivatives 21.

Figure 5: a) Structure of triflate derivatives 22. b) Stereochemistry outcomes of the reaction starting from (...

Scheme 7: Solvolysis reaction of naphthalene and anthracenyl derivatives 26 and 29.

Figure 6: Structure of bisarylated derivatives 34.

Figure 7: Structure of bisarylated derivatives 36.

Scheme 8: Reactivity of 9c in the presence of a Brønsted acid.

Scheme 9: Cationic electrocyclization of 38a–c under strongly acidic conditions.

Scheme 10: Brønsted acid-catalyzed synthesis of indenes 42 and indanes 43.

Scheme 11: Reactivity of sulfurane 44 in triflic acid.

Scheme 12: Solvolysis of triflate 45f in alcoholic solvents.

Scheme 13: Synthesis of labeled 18O-52.

Scheme 14: Reactivity of sulfurane 53 in triflic acid.

Figure 8: Structure of tosylates 56 and 21f.

Scheme 15: Resonance forms in benzylic carbenium ions.

Figure 9: Structure of pyrrole derivatives 58 and 59.

Scheme 16: Resonance structure 60↔60’.

Scheme 17: Ga(OTf)3-catalyzed synthesis of 3,3’- and 3,6’-bis(indolyl)methane from trifluoromethylated 3-indol...

Scheme 18: Proposed reaction mechanism.

Scheme 19: Metal-free 1,2-phosphorylation of 3-indolylmethanols.

Scheme 20: Superacid-mediated arylation of thiophene derivatives.

Scheme 21: In situ mechanistic NMR investigations.

Scheme 22: Proposed mechanisms for the prenyltransferase-catalyzed condensation.

Scheme 23: Influence of a CF3 group on the allylic SN1- and SN2-mechanism-based reactions.

Scheme 24: Influence of the CF3 group on the condensation reaction.

Scheme 25: Solvolysis of 90 in TFE.

Scheme 26: Solvolysis of allyl triflates 94 and 97 and isomerization attempt of 96.

Scheme 27: Proposed mechanism for the formation of 95.

Scheme 28: Formation of α-(trifluoromethyl)allylcarbenium ion 100 in a superacid.

Scheme 29: Lewis acid activation of CF3-substituted allylic alcohols.

Scheme 30: Bimetallic-cluster-stabilized α-(trifluoromethyl)carbenium ions.

Scheme 31: Reactivity of cluster-stabilized α-(trifluoromethyl)carbenium ions.

Scheme 32: α-(Trifluoromethyl)propargylium ion 122↔122’ generated from silyl ether 120 in a superacid.

Scheme 33: Formation of α-(trifluoromethyl)propargylium ions from CF3-substituted propargyl alcohols.

Scheme 34: Direct NMR observation of the protonation of some trifluoromethyl ketones in situ and the correspon...

Scheme 35: Selected resonance forms in protonated fluoroketone derivatives.

Scheme 36: Acid-catalyzed Friedel–Crafts reactions of trifluoromethyl ketones 143a,b and 147a–c.

Scheme 37: Enantioselective hydroarylation of CF3-substituted ketones.

Scheme 38: Acid-catalyzed arylation of ketones 152a–c.

Scheme 39: Reactivity of 156 in a superacid.

Scheme 40: Reactivity of α-CF3-substituted heteroaromatic ketones and alcohols as well as 1,3-diketones.

Scheme 41: Reactivity of 168 with benzene in the presence of a Lewis or Brønsted acid.

Scheme 42: Acid-catalyzed three-component asymmetric reaction.

Scheme 43: Anodic oxidation of amines 178a–c and proposed mechanism.

Scheme 44: Reactivity of 179b in the presence of a strong Lewis acid.

Scheme 45: Trifluoromethylated derivatives as precursors of trifluoromethylated iminium ions.

Scheme 46: Mannich reaction with trifluoromethylated hemiaminal 189.

Scheme 47: Suitable nucleophiles reacting with 192 after Lewis acid activation.

Scheme 48: Strecker reaction involving the trifluoromethylated iminium ion 187.

Scheme 49: Reactivity of 199 toward nucleophiles.

Scheme 50: Reactivity of 204a with benzene in the presence of a Lewis acid.

Scheme 51: Reactivity of α-(trifluoromethyl)-α-chloro sulfides in the presence of strong Lewis acids.

Scheme 52: Anodic oxidation of sulfides 213a–h and Pummerer rearrangement.

Scheme 53: Mechanism for the electrochemical oxidation of the sulfide 213a.

Scheme 54: Reactivity of (trifluoromethyl)diazomethane (217a) in HSO3F.

Figure 10: a) Structure of diazoalkanes 217a–c and b) rate-limiting steps of their decomposition.

Scheme 55: Deamination reaction of racemic 221 and enantioenriched (S)-221.

Scheme 56: Deamination reaction of labeled 221-d2. Elimination products were formed in this reaction, the yiel...

Scheme 57: Deamination reaction of 225-d2. Elimination products were also formed in this reaction in undetermi...

Scheme 58: Formation of 229 from 228 via 1,2-H-shift.

Scheme 59: Deamination reaction of 230. Elimination products were formed in this reaction, the yield of which ...

Scheme 60: Deamination of several diazonium ions. Elimination products were formed in these reactions, the yie...

Scheme 61: Solvolysis reaction mechanism of alkyl tosylates.

Scheme 62: Solvolysis outcome for the tosylates 248 and 249 in HSO3FSbF5.

Figure 11: Solvolysis rate of 248, 249, 252, and 253 in 91% H2SO4.

Scheme 63: Illustration of the reaction pathways. TsCl, pyridine, −5 °C (A); 98% H2SO4, 30 °C (B); 98% H2SO4, ...

Scheme 64: Proposed solvolysis mechanism for the aliphatic tosylate 248.

Scheme 65: Solvolysis of the derivatives 259 and 260.

Scheme 66: Solvolysis of triflate 261. SOH = solvent.

Scheme 67: Intramolecular Friedel–Crafts alkylations upon the solvolysis of triflates 264 and 267.

Scheme 68: α-CF3-enhanced γ-silyl elimination of cyclobutyltosylates 270a,b.

Scheme 69: γ-Silyl elimination in the synthesis of a large variety of CF3-substituted cyclopropanes. Pf = pent...

Scheme 70: Synthetic pathways to 281. aNMR yields.

Scheme 71: The cyclopropyl-substituted homoallylcyclobutylcarbenium ion manifold.

Scheme 72: Reactivity of CF3-substituted cyclopropylcarbinyl derivatives 287a–c. LG = leaving group.

Scheme 73: Reactivity of CF3-substituted cyclopropylcarbinyl derivatives 291a–c.

Scheme 74: Superacid-promoted dimerization or TFP.

Scheme 75: Reactivity of TFP in a superacid.

Scheme 76: gem-Difluorination of α-fluoroalkyl styrenes via the formation of a “hidden” α-RF-substituted carbe...

Scheme 77: Solvolysis of CF3-substituted pentyne 307.

Scheme 78: Photochemical rearrangement of 313.

Figure 12: Structure of 2-norbornylcarbenium ion 318 and argued model for the stabilization of this cation.

Figure 13: Structures and solvolysis rate (TFE, 25 °C) of the sulfonates 319–321. Mos = p-MeOC6H4SO2.

Scheme 79: Mechanism for the solvolysis of 323. SOH = solvent.

Scheme 80: Products formed by the hydrolysis of 328.

Scheme 81: Proposed carbenium ion intermediates in an equilibrium during the solvolysis of tosylates 328, 333,...
The preparation and properties of 1,1-difluorocyclopropane derivatives
- Kymbat S. Adekenova,
- Peter B. Wyatt and
- Sergazy M. Adekenov
Beilstein J. Org. Chem. 2021, 17, 245–272, doi:10.3762/bjoc.17.25

- opposite to the CF2 moiety, which was followed by the recyclization of the intermediate diradical (Scheme 42). The activation energy for the rearrangement of 90 was lower by 9.4 kcal/mol than for the parent hydrocarbon system 92. The activation energy of the trans-isomer 91 was greater than that of cis

Graphical Abstract

Scheme 1: Synthesis of 1,1-difluoro-2,3-dimethylcyclopropane (2).

Scheme 2: Cyclopropanation via dehydrohalogenation of chlorodifluoromethane.

Scheme 3: Difluorocyclopropanation of methylstyrene 7 using dibromodifluoromethane and zinc.

Scheme 4: Synthesis of difluorocyclopropanes from the reaction of dibromodifluoromethane and triphenylphosphi...

Scheme 5: Generation of difluorocarbene in a catalytic two-phase system and its addition to tetramethylethyle...

Scheme 6: The reaction of methylstyrene 7 with chlorodifluoromethane (11) in the presence of a tetraarylarson...

Scheme 7: Pyrolysis of sodium chlorodifluoroacetate (12) in refluxing diglyme in the presence of alkene 13.

Scheme 8: Synthesis of boron-substituted gem-difluorocyclopropanes 16.

Scheme 9: Addition of sodium bromodifluoroacetate (17) to alkenes.

Scheme 10: Addition of sodium bromodifluoroacetate (17) to silyloxy-substituted cyclopropanes 20.

Scheme 11: Synthesis of difluorinated nucleosides.

Scheme 12: Addition of butyl acrylate (26) to difluorocarbene generated from TFDA (25).

Scheme 13: Addition of difluorocarbene to propargyl esters 27 and conversion of the difluorocyclopropenes 28 t...

Scheme 14: The generation of difluorocyclopropanes using MDFA 30.

Scheme 15: gem-Difluorocyclopropanation of styrene (32) using difluorocarbene generated from TMSCF3 (31) under...

Scheme 16: Synthesis of a gem-difluorocyclopropane derivative using HFPO (41) as a source of difluorocarbene.

Scheme 17: Cyclopropanation of (Z)-2-butene in the presence of difluorodiazirine (44).

Scheme 18: The cyclopropanation of 1-octene (46) using Seyferth's reagent (45) as a source of difluorocarbene.

Scheme 19: Alternative approaches for the difluorocarbene synthesis from trimethyl(trifluoromethyl)tin (48).

Scheme 20: Difluorocyclopropanation of cyclohexene (49).

Scheme 21: Synthesis of difluorocyclopropane derivative 53 using bis(trifluoromethyl)cadmium (51) as the diflu...

Scheme 22: Addition of difluorocarbene generated from tris(trifluoromethyl)bismuth (54).

Scheme 23: Addition of a stable (trifluoromethyl)zinc reagent to styrenes.

Scheme 24: The preparation of 2,2-difluorocyclopropanecarboxylic acids of type 58.

Scheme 25: Difluorocyclopropanation via Michael cyclization.

Scheme 26: Difluorocyclopropanation using N-acylimidazolidinone 60.

Scheme 27: Difluorocyclopropanation through the cyclization of phenylacetonitrile (61) and 1,2-dibromo-1,1-dif...

Scheme 28: gem-Difluoroolefins 64 for the synthesis of functionalized cyclopropanes 65.

Scheme 29: Preparation of aminocyclopropanes 70.

Scheme 30: Synthesis of fluorinated methylenecyclopropane 74 via selenoxide elimination.

Scheme 31: Reductive dehalogenation of (1R,3R)-75.

Scheme 32: Synthesis of chiral monoacetates by lipase catalysis.

Scheme 33: Transformation of (±)-trans-81 using Rhodococcus sp. AJ270.

Scheme 34: Transformation of (±)-trans-83 using Rhodococcus sp. AJ270.

Scheme 35: Hydrogenation of difluorocyclopropenes through enantioselective hydrocupration.

Scheme 36: Enantioselective transfer hydrogenation of difluorocyclopropenes with a Ru-based catalyst.

Scheme 37: The thermal transformation of trans-1,2-dichloro-3,3-difluorocyclopropane (84).

Scheme 38: cis–trans-Epimerization of 1,1-difluoro-2,3-dimethylcyclopropane.

Scheme 39: 2,2-Difluorotrimethylene diradical intermediate.

Scheme 40: Ring opening of stereoisomers 88 and 89.

Scheme 41: [1,3]-Rearrangement of alkenylcyclopropanes 90–92.

Scheme 42: Thermolytic rearrangement of 2,2-difluoro-1-vinylcyclopropane (90).

Scheme 43: Thermal rearrangement for ethyl 3-(2,2-difluoro)-3-phenylcyclopropyl)acrylates 93 and 95.

Scheme 44: Possible pathways of the ring opening of 1,1-difluoro-2-vinylcyclopropane.

Scheme 45: Equilibrium between 1,1-difluoro-2-methylenecyclopropane (96) and (difluoromethylene)cyclopropane 97...

Scheme 46: Ring opening of substituted 1,1-difluoro-2,2-dimethyl-3-methylenecyclopropane 98.

Scheme 47: 1,1-Difluorospiropentane rearrangement.

Scheme 48: Acetolysis of (2,2-difluorocyclopropyl)methyl tosylate (104) and (1,1-difluoro-2-methylcyclopropyl)...

Scheme 49: Ring opening of gem-difluorocyclopropyl ketones 106 and 108 by thiolate nucleophiles.

Scheme 50: Hydrolysis of gem-difluorocyclopropyl acetals 110.

Scheme 51: Ring-opening reaction of 2,2-difluorocyclopropyl ketones 113 in the presence of ionic liquid as a s...

Scheme 52: Ring opening of gem-difluorocyclopropyl ketones 113a by MgI2-initiated reaction with diarylimines 1...

Scheme 53: Ring-opening reaction of gem-difluorocyclopropylstannanes 117.

Scheme 54: Preparation of 1-fluorovinyl vinyl ketone 123 and the synthesis of 2-fluorocyclopentenone 124. TBAT...

Scheme 55: Iodine atom-transfer ring opening of 1,1-difluoro-2-(1-iodoalkyl)cyclopropanes 125a–c.

Scheme 56: Ring opening of bromomethyl gem-difluorocyclopropanes 130 and formation of gem-difluoromethylene-co...

Scheme 57: Ring-opening aerobic oxidation reaction of gem-difluorocyclopropanes 132.

Scheme 58: Dibrominative ring-opening functionalization of gem-difluorocyclopropanes 134.

Scheme 59: The selective formation of (E,E)- and (E,Z)-fluorodienals 136 and 137 from difluorocyclopropyl acet...

Scheme 60: Proposed mechanism for the reaction of difluoro(methylene)cyclopropane 139 with Br2.

Scheme 61: Thermal rearrangement of F2MCP 139 and iodine by CuI catalysis.

Scheme 62: Synthesis of 2-fluoropyrroles 142.

Scheme 63: Ring opening of gem-difluorocyclopropyl ketones 143 mediated by BX3.

Scheme 64: Lewis acid-promoted ring-opening reaction of 2,2-difluorocyclopropanecarbonyl chloride (148).

Scheme 65: Ring-opening reaction of the gem-difluorocyclopropyl ketone 106 by methanolic KOH.

Scheme 66: Hydrogenolysis of 1,1-difluoro-3-methyl-2-phenylcyclopropane (151).

Scheme 67: Synthesis of monofluoroalkenes 157.

Scheme 68: The stereoselective Ag-catalyzed defluorinative ring-opening diarylation of 1-trimethylsiloxy-2,2-d...

Scheme 69: Synthesis of 2-fluorinated allylic compounds 162.

Scheme 70: Pd-catalyzed cross-coupling reactions of gem-difluorinated cyclopropanes 161.

Scheme 71: The (Z)-selective Pd-catalyzed ring-opening sulfonylation of 2-(2,2-difluorocyclopropyl)naphthalene...

Figure 1: Structures of zosuquidar hydrochloride and PF-06700841.

Scheme 72: Synthesis of methylene-gem-difluorocyclopropane analogs of nucleosides.

Figure 2: Anthracene-difluorocyclopropane hybrid derivatives.

Figure 3: Further examples of difluorcyclopropanes in modern drug discovery.
Pentannulation of N-heterocycles by a tandem gold-catalyzed [3,3]-rearrangement/Nazarov reaction of propargyl ester derivatives: a computational study on the crucial role of the nitrogen atom
- Giovanna Zanella,
- Martina Petrović,
- Dina Scarpi,
- Ernesto G. Occhiato and
- Enrique Gómez-Bengoa
Beilstein J. Org. Chem. 2020, 16, 3059–3068, doi:10.3762/bjoc.16.255

- species to the alkyne 5, as in I (Figure 2). The first step TS1 has a low activation energy (ΔG‡ = 12.2 kcal⋅mol−1) to form the unstable cyclic intermediate II. This short-lived species rapidly reopens through TS2 (ΔΔG‡ = 8.1 kcal⋅mol−1) to give the pentadienyl cation III, which presents a high stability
- derivative 1 (Figure 3), confirming the differences that the 2- and 3-substitution, respectively, exert in the reaction outcome. Starting with V, the acetate rearrangement is rate determining (ΔΔG‡ = 14.2 kcal⋅mol−1), and more importantly, the activation energy for the cyclization in TS6 is very low (ΔΔG
- conjugation. However, according to the energy profile, this observation does not have a reflection in the deprotonation step, which seems to be affected partially by the steric hindrance around the two hydrogen atoms, being clearly higher in Ha (a 2.2 kcal⋅mol−1 higher activation energy of TS10 than for TS8

Graphical Abstract

Figure 1: Tandem acetate rearrangement/Nazarov cyclization of different substrates.

Figure 2: DFT-computed energy profile of the tandem Au(I)-catalyzed [3,3]-rearrangement/Nazarov reaction of 3...

Figure 3: DFT-computed energy profile of the tandem Au(I)-catalyzed [3,3]-rearrangement/Nazarov reaction of 2...

Figure 4: Computed comparison of the NBO charges of 2- and 3-substituted substrates.

Figure 5: Single-step transformation of IV to IX.

Figure 6: Triflate-promoted hydrogen abstraction and protodeauration with HOTf.

Figure 7: Triflate-mediated abstraction of the hydrogen atom Ha and protodeauration.

Scheme 1: Synthesis of the enynyl acetate starting material 14.

Scheme 2: Synthesis and cyclization of enynyl acetate 20.
Dirhamnolipid ester – formation of reverse wormlike micelles in a binary (primerless) system
- David Liese,
- Hans Henning Wenk,
- Xin Lu,
- Jochen Kleinen and
- Gebhard Haberhauer
Beilstein J. Org. Chem. 2020, 16, 2820–2830, doi:10.3762/bjoc.16.232

- contour length decreases with increasing temperature according to the following Arrhenius equations, Equation 9 and Equation 10 [32]: where Ea is the activation energy, R is the gas constant, and A is the preexponential factor. The semilogarithmic plots of η0 and τR versus 1/T (Figure 7b and c) indicate
- an Arrhenius plot like behavior, and the activation energy calculated from the slopes of the two plots equals 119 kJ/mol, close to those found in other wormlike micelles [48][49][50][51][52]. According to Equation 10, G0 is independent of the temperature, which is not true in our case. This might be

Graphical Abstract

Figure 1: Chemical structure of dirhamnolipid 1.

Scheme 1: Synthesis of the dirhamnolipid esters and the chemical structure of 7.

Figure 2: Solubility of the dirhamnolipid esters in various solvents (+ = soluble, − = insoluble, G = gel).

Figure 3: Phase transition temperature for the dirhamnolipid esters in toluene while heating (TGS, blue) and ...

Figure 4: Amplitude sweep: double logarithmic plot of the dynamic moduli against the amplitude (deformation) ...

Figure 5: Frequency sweep: double logarithmic plot of the dynamic moduli against the frequency for the dirham...

Figure 6: Double logarithmic plot of (a) the plateau modulus G0 and (b) the relaxation time τR against the co...

Figure 7: Semilogarithmic plot of (a) G0/G''min, (b) η0, and (c) τR against the inverse absolute temperature ...

Figure 8: Polarized optical microscopy (POM) images of the 2/toluene system (5 wt %) with crossed polarizers ...

Figure 9: Schematic representation of the formation of RWLM by dirhamnolipid esters.
Recent developments in enantioselective photocatalysis
- Callum Prentice,
- James Morrisson,
- Andrew D. Smith and
- Eli Zysman-Colman
Beilstein J. Org. Chem. 2020, 16, 2363–2441, doi:10.3762/bjoc.16.197

- photocatalysts (PCs) generate excited state substrates that can then undergo reactions that would be impossible in the ground state [4]. A challenge for enantioselective catalysis is stifling the racemic background reaction, which is generally achieved through a lower activation energy for the catalysed process

Graphical Abstract

Scheme 1: Amine/photoredox-catalysed α-alkylation of aldehydes with alkyl bromides bearing electron-withdrawi...

Scheme 2: Amine/HAT/photoredox-catalysed α-functionalisation of aldehydes using alkenes.

Scheme 3: Amine/cobalt/photoredox-catalysed α-functionalisation of ketones and THIQs.

Scheme 4: Amine/photoredox-catalysed α-functionalisation of aldehydes or ketones with imines. (a) Using keton...

Scheme 5: Bifunctional amine/photoredox-catalysed enantioselective α-functionalisation of aldehydes.

Scheme 6: Bifunctional amine/photoredox-catalysed α-functionalisation of aldehydes using amine catalysts via ...

Scheme 7: Amine/photoredox-catalysed RCA of iminium ion intermediates. (a) Synthesis of quaternary stereocent...

Scheme 8: Bifunctional amine/photoredox-catalysed RCA of enones in a radical chain reaction initiated by an i...

Scheme 9: Bifunctional amine/photoredox-catalysed RCA reactions of iminium ions with different radical precur...

Scheme 10: Bifunctional amine/photoredox-catalysed radical cascade reactions between enones and alkenes with a...

Scheme 11: Amine/photocatalysed photocycloadditions of iminium ion intermediates. (a) External photocatalyst u...

Scheme 12: Amine/photoredox-catalysed addition of acrolein (94) to iminium ions.

Scheme 13: Dual NHC/photoredox-catalysed acylation of THIQs.

Scheme 14: NHC/photocatalysed spirocyclisation via photoisomerisation of an extended Breslow intermediate.

Scheme 15: CPA/photoredox-catalysed aza-pinacol cyclisation.

Scheme 16: CPA/photoredox-catalysed Minisci-type reaction between azaarenes and α-amino radicals.

Scheme 17: CPA/photoredox-catalysed radical additions to azaarenes. (a) α-Amino radical or ketyl radical addit...

Scheme 18: CPA/photoredox-catalysed reduction of azaarene-derived substrates. (a) Reduction of ketones. (b) Ex...

Scheme 19: CPA/photoredox-catalysed radical coupling reactions of α-amino radicals with α-carbonyl radicals. (...

Scheme 20: CPA/photoredox-catalysed Povarov reaction.

Scheme 21: CPA/photoredox-catalysed reactions with imines. (a) Decarboxylative imine generation followed by Po...

Scheme 22: Bifunctional CPA/photocatalysed [2 + 2] photocycloadditions.

Scheme 23: PTC/photocatalysed oxygenation of 1-indanone-derived β-keto esters.

Scheme 24: PTC/photoredox-catalysed perfluoroalkylation of 1-indanone-derived β-keto esters via a radical chai...

Scheme 25: Bifunctional hydrogen bonding/photocatalysed intramolecular [2 + 2] photocycloadditions of quinolon...

Scheme 26: Bifunctional hydrogen bonding/photocatalysed intramolecular RCA cyclisation of a quinolone.

Scheme 27: Bifunctional hydrogen bonding/photocatalysed intramolecular [2 + 2] photocycloadditions of quinolon...

Scheme 28: Bifunctional hydrogen bonding/photocatalysed [2 + 2] photocycloaddition reactions. (a) First use of...

Scheme 29: Bifunctional hydrogen bonding/photocatalysed deracemisation of allenes.

Scheme 30: Bifunctional hydrogen bonding/photocatalysed deracemisation reactions. (a) Deracemisation of sulfox...

Scheme 31: Bifunctional hydrogen bonding/photocatalysed intramolecular [2 + 2] photocycloaddition of coumarins....

Scheme 32: Bifunctional hydrogen bonding/photocatalysed [2 + 2] photocycloadditions of quinolones. (a) Intramo...

Scheme 33: Hydrogen bonding/photocatalysed formal arylation of benzofuranones.

Scheme 34: Hydrogen bonding/photoredox-catalysed dehalogenative protonation of α,α-chlorofluoro ketones.

Scheme 35: Hydrogen bonding/photoredox-catalysed reductions. (a) Reduction of 1,2-diketones. (b) Reduction of ...

Scheme 36: Hydrogen bonding/HAT/photocatalysed deracemisation of cyclic ureas.

Scheme 37: Hydrogen bonding/HAT/photoredox-catalysed synthesis of cyclic sulfonamides.

Scheme 38: Hydrogen bonding/photoredox-catalysed reaction between imines and indoles.

Scheme 39: Chiral cation/photoredox-catalysed radical coupling of two α-amino radicals.

Scheme 40: Chiral phosphate/photoredox-catalysed hydroetherfication of alkenols.

Scheme 41: Chiral phosphate/photoredox-catalysed synthesis of pyrroloindolines.

Scheme 42: Chiral anion/photoredox-catalysed radical cation Diels–Alder reaction.

Scheme 43: Lewis acid/photoredox-catalysed cycloadditions of carbonyls. (a) Formal [2 + 2] cycloaddition of en...

Scheme 44: Lewis acid/photoredox-catalysed RCA reaction using a scandium Lewis acid between α-amino radicals a...

Scheme 45: Lewis acid/photoredox-catalysed RCA reaction using a copper Lewis acid between α-amino radicals and...

Scheme 46: Lewis acid/photoredox-catalysed synthesis of 1,2-amino alcohols from aldehydes and nitrones using a...

Scheme 47: Lewis acid/photocatalysed [2 + 2] photocycloadditions of enones and alkenes.

Scheme 48: Meggers’s chiral-at-metal catalysts.

Scheme 49: Lewis acid/photoredox-catalysed α-functionalisation of ketones with alkyl bromides bearing electron...

Scheme 50: Bifunctional Lewis acid/photoredox-catalysed radical coupling reaction using α-chloroketones and α-...

Scheme 51: Lewis acid/photocatalysed RCA of enones. (a) Using aldehydes as acyl radical precursors. (b) Other ...

Scheme 52: Bifunctional Lewis acid/photocatalysis for a photocycloaddition of enones.

Scheme 53: Lewis acid/photoredox-catalysed RCA reactions of enones using DHPs as radical precursors.

Scheme 54: Lewis acid/photoredox-catalysed functionalisation of β-ketoesters. (a) Hydroxylation reaction catal...

Scheme 55: Bifunctional copper-photocatalysed alkylation of imines.

Scheme 56: Copper/photocatalysed alkylation of imines. (a) Bifunctional copper catalysis using α-silyl amines....

Scheme 57: Bifunctional Lewis acid/photocatalysed intramolecular [2 + 2] photocycloaddition.

Scheme 58: Bifunctional Lewis acid/photocatalysed [2 + 2] photocycloadditions (a) Intramolecular cycloaddition...

Scheme 59: Bifunctional Lewis acid/photocatalysed rearrangement of 2,4-dieneones.

Scheme 60: Lewis acid/photocatalysed [2 + 2] cycloadditions of cinnamate esters and styrenes.

Scheme 61: Nickel/photoredox-catalysed arylation of α-amino acids using aryl bromides.

Scheme 62: Nickel/photoredox catalysis. (a) Desymmetrisation of cyclic meso-anhydrides using benzyl trifluorob...

Scheme 63: Nickel/photoredox catalysis for the acyl-carbamoylation of alkenes with aldehydes using TBADT as a ...

Scheme 64: Bifunctional copper/photoredox-catalysed C–N coupling between α-chloro amides and carbazoles or ind...

Scheme 65: Bifunctional copper/photoredox-catalysed difunctionalisation of alkenes with alkynes and alkyl or a...

Scheme 66: Copper/photoredox-catalysed decarboxylative cyanation of benzyl phthalimide esters.

Scheme 67: Copper/photoredox-catalysed cyanation reactions using TMSCN. (a) Propargylic cyanation (b) Ring ope...

Scheme 68: Palladium/photoredox-catalysed allylic alkylation reactions. (a) Using alkyl DHPs as radical precur...

Scheme 69: Manganese/photoredox-catalysed epoxidation of terminal alkenes.

Scheme 70: Chromium/photoredox-catalysed allylation of aldehydes.

Scheme 71: Enzyme/photoredox-catalysed dehalogenation of halolactones.

Scheme 72: Enzyme/photoredox-catalysed dehalogenative cyclisation.

Scheme 73: Enzyme/photoredox-catalysed reduction of cyclic imines.

Scheme 74: Enzyme/photocatalysed enantioselective reduction of electron-deficient alkenes as mixtures of (E)/(Z...

Scheme 75: Enzyme/photoredox catalysis. (a) Deacetoxylation of cyclic ketones. (b) Reduction of heteroaromatic...

Scheme 76: Enzyme/photoredox-catalysed synthesis of indole-3-ones from 2-arylindoles.

Scheme 77: Enzyme/HAT/photoredox catalysis for the DKR of primary amines.

Scheme 78: Bifunctional enzyme/photoredox-catalysed benzylic C–H hydroxylation of trifluoromethylated arenes.
Photosensitized direct C–H fluorination and trifluoromethylation in organic synthesis
- Shahboz Yakubov and
- Joshua P. Barham
Beilstein J. Org. Chem. 2020, 16, 2151–2192, doi:10.3762/bjoc.16.183

- reactions. Keywords: C–H activation; energy transfer; fluorination; photocatalysis; photosensitization; visible light; Review 1 Introduction 1.1 Importance of direct C–H fluorination/trifluoromethylation and photosensitization in organic synthesis 1.1.1 Importance of fluorine atoms in organic molecules
- electrophilic fluorinating source. Thereafter, the AQN–Selectfluor® exciplex abstracts a hydrogen atom from the C3 position of pentane to form a secondary radical, a Selectfluor® N-radical cation, HF and AQN. The activation energy barrier relative to RC2 was only 9.9 kcal⋅mol−1 (Scheme 17). The Selectfluor® N
- -radical cation can abstract a hydrogen atom from the C3 2° C–H bond of another pentane molecule in an overall exergonic process with an activation energy barrier of 2.0 kcal⋅mol−1 from Int4 to afford Int5 and ultimately Int6 (Scheme 18). The authors did not mention the possibility of a mechanism whereby

Graphical Abstract

Figure 1: Fluorine-containing drugs.

Figure 2: Fluorinated agrochemicals.

Scheme 1: Selectivity of fluorination reactions.

Scheme 2: Different mechanisms of photocatalytic activation. Sub = substrate.

Figure 3: Jablonski diagram showing visible-light-induced energy transfer pathways: a) absorption, b) IC, c) ...

Figure 4: Schematic illustration of TTET.

Figure 5: Organic triplet PSCats.

Figure 6: Additional organic triplet PSCats.

Figure 7: A) Further organic triplet PSCats and B) transition metal triplet PSCats.

Figure 8: Different fluorination reagents grouped by generation.

Scheme 3: Synthesis of Selectfluor®.

Scheme 4: General mechanism of PS TTET C(sp3)–H fluorination.

Scheme 5: Selective benzylic mono- and difluorination using 9-fluorenone and xanthone PSCats, respectively.

Scheme 6: Chen’s photosensitized monofluorination: reaction scope.

Scheme 7: Chen’s photosensitized benzylic difluorination reaction scope.

Scheme 8: Photosensitized monofluorination of ethylbenzene on a gram scale.

Scheme 9: Substrate scope of Tan’s AQN-photosensitized C(sp3)–H fluorination.

Scheme 10: AQN-photosensitized C–H fluorination reaction on a gram scale.

Scheme 11: Reaction mechanism of the AQN-assisted fluorination.

Figure 9: 3D structures of the singlet ground and triplet excited states of Selectfluor®.

Scheme 12: Associated transitions for the activation of acetophenone by violet light.

Scheme 13: Ethylbenzene C–H fluorination with various PSCats and conditions.

Scheme 14: Effect of different PSCats on the C(sp3)–H fluorination of cyclohexane (39).

Scheme 15: Reaction scope of Chen’s acetophenone-photosensitized C(sp3)–H fluorination reaction.

Figure 10: a) Site-selectivity of Chen’s acetophenone-photosensitized C–H fluorination reaction [201]. b) Site-sele...

Scheme 16: Formation of the AQN–Selectfluor® exciplex Int1.

Scheme 17: Generation of the C3 2° pentane radical and the Selectfluor® N-radical cation from the exciplex.

Scheme 18: Hydrogen atom abstraction by the Selectfluor® N-radical cation from pentane to give the C3 2° penta...

Scheme 19: Fluorine atom transfer from Selectfluor® to the C3 2° pentane radical to yield 3-fluoropentane and ...

Scheme 20: Barrierless fluorine atom transfer from Int1 to the C3 2° pentane radical to yield 3-fluoropentane,...

Scheme 21: Ketone-directed C(sp3)–H fluorination.

Scheme 22: Ketone-directed fluorination through a 5- and a 6-membered transition state, respectively.

Scheme 23: Effect of different PSCats on the photosensitized C(sp3)–H fluorination of 47.

Scheme 24: Substrate scope of benzil-photoassisted C(sp3)–H fluorinations.

Scheme 25: A) Benzil-photoassisted enone-directed C(sp3)–H fluorination. B) Classification of the reaction mod...

Scheme 26: A) Xanthone-photoassisted ketal-directed C(sp3)–H fluorination. B) Substrate scope. C) C–H fluorina...

Scheme 27: Rationale for the selective HAT at the C2 C–H bond of galactose acetonide.

Scheme 28: Photosensitized C(sp3)–H benzylic fluorination of a peptide using different PSCats.

Scheme 29: Peptide scope of 5-benzosuberenone-photoassisted C(sp3)–H fluorinations.

Scheme 30: Continuous flow PS TTET monofluorination of 72.

Scheme 31: Photosensitized C–H fluorination of N-butylphthalimide as a PSX.

Scheme 32: Substrate scope and limitations of the PSX C(sp3)–H monofluorination.

Scheme 33: Substrate crossover monofluorination experiment.

Scheme 34: PS TTET mechanism proposed by Hamashima and co-workers.

Scheme 35: Photosensitized TFM of 78 to afford α-trifluoromethylated ketone 80.

Scheme 36: Substrate scope for photosensitized styrene TFM to give α-trifluoromethylated ketones.

Scheme 37: Control reactions for photosensitized TFM of styrenes.

Scheme 38: Reaction mechanism for photosensitized TFM of styrenes to afford α-trifluoromethylated ketones.

Scheme 39: Reaction conditions for TFMs to yield the cis- and the trans-product, respectively.

Scheme 40: Substrate scope of trifluoromethylated (E)-styrenes.

Scheme 41: Strategies toward trifluoromethylated (Z)-styrenes.

Scheme 42: Substrate scope of trifluoromethylated (Z)-styrenes.

Scheme 43: Reaction mechanism for photosensitized TFM of styrenes to afford E- or Z-products.
One-pot synthesis of oxazolidinones and five-membered cyclic carbonates from epoxides and chlorosulfonyl isocyanate: theoretical evidence for an asynchronous concerted pathway
- Esra Demir,
- Ozlem Sari,
- Yasin Çetinkaya,
- Ufuk Atmaca,
- Safiye Sağ Erdem and
- Murat Çelik
Beilstein J. Org. Chem. 2020, 16, 1805–1819, doi:10.3762/bjoc.16.148
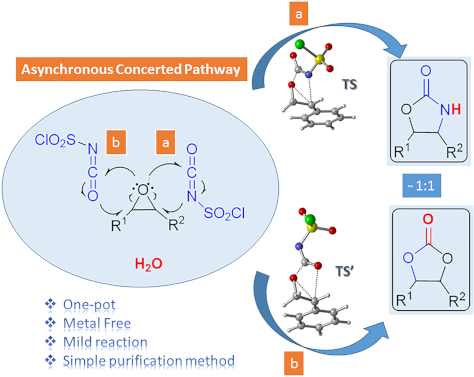
- the transition state and so lowering the activation energy barrier (Figure 3a). On the other hand, stabilization of the benzylic cation is not possible along the IRC path for TS1′ (Figure 3b), since the bond distance C2–C(Ph) is found as around 1.50 Å showing a single bond character. This can be the

Graphical Abstract

Scheme 1: Oxazolidinone (1), five-membered cyclic carbonate (2) and some important compounds containing an ox...

Scheme 2: Proposed mechanisms by Keshava Murthy and Dhar [41] and De Meijere and co-workers [42].

Figure 1: Possible pathways for the formation of oxazolidinone intermediates 10 and 11. Optimized transition ...

Figure 2: Potential energy profile related to the formation of oxazolidinone intermediates 10 and 11 at the P...

Figure 3: IRC calculated for the formation of (a) 10 and (b) 11 at M06-2X/6-31+G(d,p) level. I-1, I-15, I-35, ...

Figure 4: Optimized geometries for the stationary points for the formation of 10 at PCM(DCM)/M06-2X/6-31+G(d,...

Scheme 3: Proposed mechanisms for the formation of oxazolidinone 9f.

Figure 5: Potential energy profiles for paths 1a (blue), 1b (red), 2 (green) and relative Gibbs free energies...

Figure 6: Optimized geometries for the stationary points of path 1b at PCM(DCM)/M06-2X/6-31+G(d,p)//M06-2X/6-...

Scheme 4: Proposed mechanism for the formation of five-membered cyclic carbonate 8f.

Figure 7: Potential energy profile and relative Gibbs free energies (kcal/mol) in DCM related to the formatio...

Figure 8: Optimized geometries for the stationary points of step 1 for the formation of 16 at PCM(DCM)/M06-2X...

Figure 9: Optimized geometries for the stationary points of step 2 for the formation of 17 at PCM(DCM)/M06-2X...

Figure 10: Optimized geometries for the stationary points of step 3 for the formation of PC8 at PCM(DCM)/M06-2...
p-Pyridinyl oxime carbamates: synthesis, DNA binding, DNA photocleaving activity and theoretical photodegradation studies
- Panagiotis S. Gritzapis,
- Panayiotis C. Varras,
- Nikolaos-Panagiotis Andreou,
- Katerina R. Katsani,
- Konstantinos Dafnopoulos,
- George Psomas,
- Zisis V. Peitsinis,
- Alexandros E. Koumbis and
- Konstantina C. Fylaktakidou
Beilstein J. Org. Chem. 2020, 16, 337–350, doi:10.3762/bjoc.16.33

- corresponding activation energy (Equation 1) and free activation energy (Equation 2) were calculated for compound 12 and found 3.14 and 2.95 kcal/mol, respectively. These values were used in Equation 3 in order to calculate the rate constant for the N−O bond dissociation. Accordingly, kr, was found to be 4.27
- characterized accordingly as stationary points (minima or maxima) on the corresponding potential energy surfaces (PESs). Equations 1–5 were used for the calculations of the rates and the physicochemical data of the N−O bond dissociation of the most active compound 12 in radicals. The corresponding activation
- energy and free energy of activation are given in Equation 1 and Equation 2, respectively: For the calculation of the rate constant, kr, the Eyring’s classical Equation 3 was used, where in the above equation kB is the Boltzmann’s constant (1.380662∙10−23 J/K), h is the Planck’s constant (6.626176∙10−34

Graphical Abstract

Figure 1: General structures of oxime derivatives with possible DNA photocleavage ability. Left: Oxime carbox...

Scheme 1: Synthesis of O-carbamoyl amidoximes (8–13), ethanone oximes (15–20) and aldoximes (22–27). Oxime 1 ...

Figure 2: UV–vis spectra of CT DNA ([DNA] = 1.1 × 10−4 M) in buffer solution in the absence or presence of in...

Figure 3: Relative viscosity (η/η0)1/3 of CT DNA (0.1 mM) in buffer solution in the presence of compounds 11 ...

Figure 4: Plot of EB-DNA relative fluorescence emission intensity at λ = 592 nm (I/I0, %) vs r (= [compound]/...

Figure 5: DNA photocleavage of amidoxime carbamates at a concentration of 500 μM and mechanistic studies of a...

Figure 6: Potential energy curve for the dissociation of 12 in the first excited triplet state, T1. For compo...

Scheme 2: Photodissociation reaction of the derivative 12 in the T1 state and the formation of ground state r...

Scheme 3: Decarboxylation reaction of the p-chlorophenylcarbamoyloxyl radical.

Figure 7: Proposed scheme showing a possible energy transfer from acetophenone sensitizer to oxime carbamate ...

Figure 8: DNA photocleavage of compounds 8–10 and 12–13 at concentration of 500 μM, at 365 nm, in the absence...

Figure 9: DNA photocleavage of compound 12 at a concentration of 500 μM, at 312 nm, in the absence and presen...
The reaction of arylmethyl isocyanides and arylmethylamines with xanthate esters: a facile and unexpected synthesis of carbamothioates
- Narasimhamurthy Rajeev,
- Toreshettahally R. Swaroop,
- Ahmad I. Alrawashdeh,
- Shofiur Rahman,
- Abdullah Alodhayb,
- Seegehalli M. Anil,
- Kuppalli R. Kiran,
- Chandra,
- Paris E. Georghiou,
- Kanchugarakoppal S. Rangappa and
- Maralinganadoddi P. Sadashiva
Beilstein J. Org. Chem. 2020, 16, 159–167, doi:10.3762/bjoc.16.18

- been reported for other carbene hydrolyses [37][38][39]. As can be seen from Figure 5, the highest activation energy barrier is 42.2 kJ/mol. We had previously considered an alternative mechanism in which a benzylic proton is instead removed by the base [40]. For the previous mechanism, which we have
- now recalculated at the B3LYP/6-311++G(d,p) level of theory in DMF (using a PCM, see Scheme S1 and Figures S15 and S16, Supporting Information File 1), an activation energy barrier of 20.6 kJ/mol was obtained for the formation of the resulting benzylic α-carbanion and H2. This benzylic α-carbanion
- anion, either by a stepwise or by a concerted five-membered ring transition state, followed by several subsequent steps could lead to the observed products. However, the mechanism involving all of those steps required an improbably higher overall activation energy of 173.9 kJ/mol (Figure S16, Supporting

Graphical Abstract

Scheme 1: Synthesis of carbamothioates from xanthate esters and benzyl isocyanides.

Figure 1: Substrate scope for the synthesis of carbamothioates. Reaction conditions for methods A and B: sodi...

Figure 2: ORTEP diagram of O-benzyl (4-fluorobenzyl)carbamothioate (4c).

Figure 3: Rotamers of thionocarbamates 4 (top) and computer-minimized structures of 4c (bottom).

Scheme 2: Proposed general reaction mechanism for the formation of carbamothioates (e.g., 4a) from xanthate e...

Figure 4: Optimized geometries of the reactants, transition states, intermediates, and products of the propos...

Figure 5: Relative energies of the reactants, transition states (TS1–TS3), and intermediates (Int1–Int3) of t...